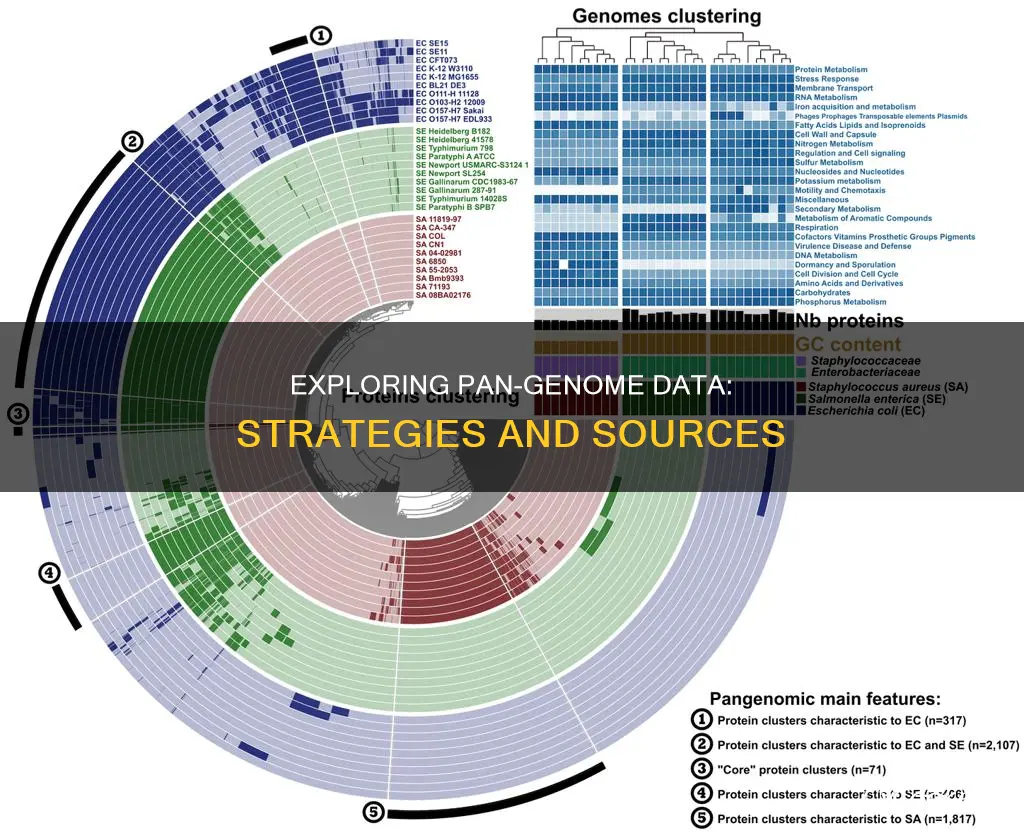
A pan-genome is the entire set of genes from all strains within a clade. The first step in a pan-genomic analysis is the homogenisation of the genome annotation. The same software should be used to annotate genomes, such as GeneMark or RAST.
Several software tools are then used to calculate the pan-genome, including BPGA, GET_HOMOLOGUES, PGAP, and Roary.
| Characteristics | Values |
|---|---|
| Definition | The pan-genome is the entire set of genes from all strains within a clade. |
| Synonyms | Pangenome, supragenome |
| Parts | Core pangenome, shell pangenome, cloud pangenome |
| Core pangenome | Contains genes present in all individuals |
| Shell pangenome | Contains genes present in two or more strains |
| Cloud pangenome | Contains genes only found in a single strain |
| Cloud pangenome synonyms | Accessory genome, dispensable genome |
| Open pangenome | The number of new gene families increases with the addition of new genomes |
| Closed pangenome | The number of new gene families does not increase with the addition of new genomes |
| Software | Prokka, Roary, BPGA, GET_HOMOLOGUES, PGAP, PanDelos, Panseq, PanX, OrthoVenn, BRIDGEcereal, Anvi'o, PPanGGOLiN, Panaroo, PGAT, EDGAR, PanTools, GET_HOMOLOGUES-EST, CamBer, Prokaryotic genome analysis tool, PanCGHweb, Micropan, FindMyFriends, Piggy, PanViz, PanVizGenerator |
Explore related products



$145.35 $66.99

$59.95 $63.99


What You'll Learn
![]()
Download the supplementary file containing information on all the strains used in the study
To get started with a pan-genome analysis, you will need to download the supplementary file containing information on all the strains used in the study. This file is usually in CSV format and can be opened using software like Microsoft Excel. The file will contain details on each strain, such as its name, genome sequence, and other relevant information.
Once you have downloaded the supplementary file, you can follow these steps to get started with your pan-genome analysis:
- Open the CSV file in Excel or a similar program.
- Review the information provided for each strain, including the strain name, genome sequence, and any other relevant details.
- Select the strains you want to include in your analysis. In some cases, you may need to download additional data or files for the selected strains.
- Save your selected strains or export them to a separate file for further analysis.
- Repeat this process for any additional strains you want to include in your pan-genome analysis.
By following these steps, you will be able to gather the necessary information on the strains used in the study and prepare them for further analysis as part of your pan-genome project.
Green Pan: Seasoning Secrets
You may want to see also
Explore related products






![]()
Open the file in Excel or similar software and select the strains
To get started with your pan-genome analysis, you will need to download the relevant data. In this case, you will be using data from a study on the core and accessory genomes of E. coli strains. The first step is to download the supplementary CSV file that contains information on all the strains used in the study. This file can be found on the ENA website or by using the ena browser tools.
Once you have downloaded the CSV file, you can open it in Excel or any other software that can open .csv files. For example, if you are using Microsoft Excel, simply double-click on the file to open it in the Excel spreadsheet. If you are using alternative software, you may need to right-click on the file and select "Open with" to choose the appropriate program.
With the file now open, you will need to select the strains you want to work with. In this case, you will be selecting 32 strains. You can do this by highlighting the relevant rows or columns in the spreadsheet. Make sure to check the file for any additional information or instructions provided by the source. It is important to carefully review the data before proceeding to ensure you have selected the correct strains and that the data is complete and consistent.
After selecting your strains, you can proceed to the next steps of your analysis. This may include downloading the selected strains, assembling and annotating the strains, and performing further analysis using specific software or tools. Remember to refer to the original source or relevant tutorials for detailed instructions on each step of the process.
Sill Pan Slope: Why It's Essential
You may want to see also
Explore related products






![]()
Download the strains from the ENA website
To download the strains from the ENA website, you can follow the steps outlined below:
First, download the supplementary csv file containing information on all the strains used in the study. You can open this file using Excel or any other software that supports .csv files. From there, select 32 strains that you wish to download. Alternatively, you can use ena browser tools, which are available as a bioconda recipe.
Once you have selected your strains, you can use the ENA File Downloader, a Java-based command-line application, to download the data for further analysis. This tool allows you to submit one or more comma-separated accessions or a file with accessions for the strains you want to download. The ENA File Downloader supports FTP or Aspera for file transfers. It also offers an easy-to-use interactive interface and can create a script that can be integrated with pipelines or run programmatically. You can download the latest version of the ENA File Downloader from ENA Tools on GitHub.
Another option for downloading strains is to use the ENA FTP Downloader, a Java GUI application also available on GitHub. With this program, you can either provide an accession to view a list of associated read or analysis files for download, or you can input a file report generated from the Advanced Search API (ENA Portal API) to perform a bulk download of files based on specific criteria.
If you prefer a more user-friendly and feature-rich interface for interacting with the FTP server, Globus is another option. Files can be downloaded through the 'EMBL-EBI Public Data' endpoint within Globus, under the '/vol1' subfolder. Globus also provides a command-line interface (CLI) that can be used without access to a graphical user interface.
For those who are comfortable with command-line tools, 'wget' and 'curl' are simple options available in Linux and Mac releases. Using either of these tools, you can download a file by simply specifying its location. For example:
> $ wget ftp://ftp.sra.ebi.ac.uk/vol1/fastq/ERR164/ERR164407/ERR164407.fastq.gz
Or
> curl -o ERR164407.fastq.gz 'ftp://ftp.sra.ebi.ac.uk/vol1/fastq/ERR164/ERR164407/ERR164407.fastq.gz'
Additionally, command-line FTP clients allow you to interactively explore the FTP server and download data to your local computer. When using an FTP client, enter 'anonymous' as the username, and press enter when prompted for a password. From there, you can use commands such as 'cd' to change directories, 'ls' to view the contents of a directory, and 'get' to specify the file you want to download.
By following these steps and utilizing the tools provided by ENA, you can efficiently download the strains you need for your pan-genome analysis.
Pans: Choosing the Right Sizes for Your Kitchen
You may want to see also
Explore related products






![]()
Use ena browser tools to download the files
The enaBrowserTools is a set of Python-based utilities that interface with the ENA web services to download data from ENA. The tools are simple to run and allow accession-based data download commands with the option to create more complex commands.
To use enaBrowserTools, you will need to have Python installed and have a basic understanding of how to use the Command Line. You can download enaBrowserTools and refer to the README file for guides on installation, set-up, and general usage.
There are two main tools for downloading data from ENA: enaDataGet and enaGroupGet. The enaDataGet tool downloads all data for a given sequence, assembly, read, or analysis accession or WGS set. The enaGroupGet tool allows you to download all data of a particular group (sequence, WGS, assembly, read, or analysis) for a given sample or study accession.
To download all data for a given accession, use the command “enaDataGet”. This works for data-holding accessions such as sequence records, assembly records, run or analysis records, or WGS sets. For example, to download the data files in the sequencing run ERR164409, use the following command:
> enaDataGet -f fastq -d
This command downloads the raw reads available in the ‘fastq’ format, which is the standardised ENA fastq format. The ‘-d’ argument specifies the destination directory for the data. If a destination directory is not provided, the data will be downloaded to the directory from which the command is run.
If you also want to download the Run metadata, you can include the “-m” argument, which will download the ENA Record in XML format:
> enaDataGet -f fastq -d
The enaGroupGet command allows you to download all data files of a certain type of Record that are associated with a Study, Sample, or given Taxon. This means you can download all raw reads in a Study or all analyses of a Sample, etc., in bulk. For example, to download all the raw read files associated with the Sample SAMEA2591084, use the following command:
> enaGroupGet -f submitted -d
This command downloads the raw reads in the ‘submitted’ format, which is the file format provided by the submitter when they provided their data to the archive. Again, you can include the “-m” argument to also download the metadata for all the resulting Runs:
> enaGroupGet -f submitted -d
Scrub Away Grease: Grill Pan Revival
You may want to see also
Explore related products






![]()
Put all the .gff files in the same folder and run Roary
To get started with creating a pan-genome of isolated genome sequences, you'll need to download the relevant genome sequences. This can be done by installing the ncbi-genome-download package, which provides several scripts to download genome sequences from NCBI FTP servers. Open a terminal and type the following command to install the package:
$ pip install ncbi-genome-download
Once the package is installed, you can download the genome assemblies you require, such as fasta sequences of all bacteria, viral genomes, RefSeq genome sequences in GenBank format, or fungal genomes. Remember, when downloading gff3 files, you need to download Genbank files with the nucleotide sequence, as gff3 files on the NCBI website contain only annotations.
Next, navigate to the directory of Roary, create a new folder, and save the downloaded sequences. You will then see multiple fasta files in the same folder.
Now, start annotating the sequences to determine the attributes and location of the genes present in them, and to obtain gff3 files, which will be used as input in Roary. This can be done using Prokka. Open the terminal and type the following commands:
$ cd Downloads/Roary/example/
$ prokka --kingdom Bacteria --outdir prokka_GCA_000006285 --genus Salmonella --locustag GCA_000006285 GCA_000006285.2_ASM828v3_genomic.fna
You can add other descriptions, such as organism details (genus, species, etc.). Ensure that you annotate all the genome sequences and remember to change the output directory name, locus tag, and assembly version accordingly. After running this command, a new directory will be created for each sequence, containing 12 files with different extensions, including the gff3 file.
Now, put all the .gff files in the same folder (e.g., ./gff) and run Roary. Copy the gff3 file from each directory into another directory. Then, open the terminal and type the following command to run Roary:
$ roary -f roary -e -n -v annotation/*.gff
At this stage, Roary will retrieve all the coding sequences, translate them into protein sequences, and generate pre-clusters. Roary will then look for paralogs using blastp and create clusters using MCL. Finally, it will take every isolate and order them according to the presence or absence of orthologs. The time taken for this process will depend on the number of sequences (or gff3 files) used.
If you want to create a pan-genome without the core alignment, use the following command:
$ roary -f ./tutorial -v ./gff_all/*.gff
If you want to change the percentage identity of blastp (not recommended to go below 90%), use the following command:
$ roary -f ./tutorial -i 90 -v ./gff_all/*.gff
These commands will result in a new directory called "tutorial" (as specified in the command), where all result files will be found. You can view the summary statistics in the 'summary_statistics.txt' file. Similarly, you will find other output files such as 'gene_presence_absence.csv', 'accessory_binary_genes.fa.newick', etc.
To visualise the results, you can use the 'roary_plots.py' script (written by Marco Galardini), which is present inside the directory named 'contrib' in the main Roary directory. Open the terminal, navigate to the tutorial directory (where all the result files are present), and type the following:
$ cd tutorial
$ /home/user/Downloads/roary/contrib/roary_plots/roary_plots.py accessory_binary_genes.fa.newick gene_presence_absence.csv
Three PNG files will be added to the tutorial directory: pangenome_frequence.png, pangenome_matrix.png, and pangenome_pie.png. Additionally, you can also visualise the Newick file in phylogeny software such as Mega for further analysis.
Shiny Roasting Pan: Bird Attractor?
You may want to see also
Frequently asked questions
A pan-genome is the entire set of genes from all strains within a clade. It can be broken down into a "core pangenome" that contains genes present in all individuals, a "shell pangenome" that contains genes present in two or more strains, and a "cloud pangenome" that contains genes only found in a single strain.
You will need a collection of isolate genome sequences in FASTA format. You will also need to annotate the genomes, which can be done using software such as GeneMark or RAST.
The first step is the homogenization of genome annotation. This involves using the same software to annotate all genomes.
There are several software tools available for pan-genomic analysis, including BPGA, GET_HOMOLOGUES, PGAP, Prokka, and Roary.
The simplest form of a pan-genome is a set of unaligned sequences, which does not provide much useful information. A better representation is a multiple sequence alignment, which provides a coordinate system with many columns specifying the particular location of genes on the pan-genome.































